PRIMUS: Pedigree Reconstruction and Identification of a Maximum Unrelated Set
|
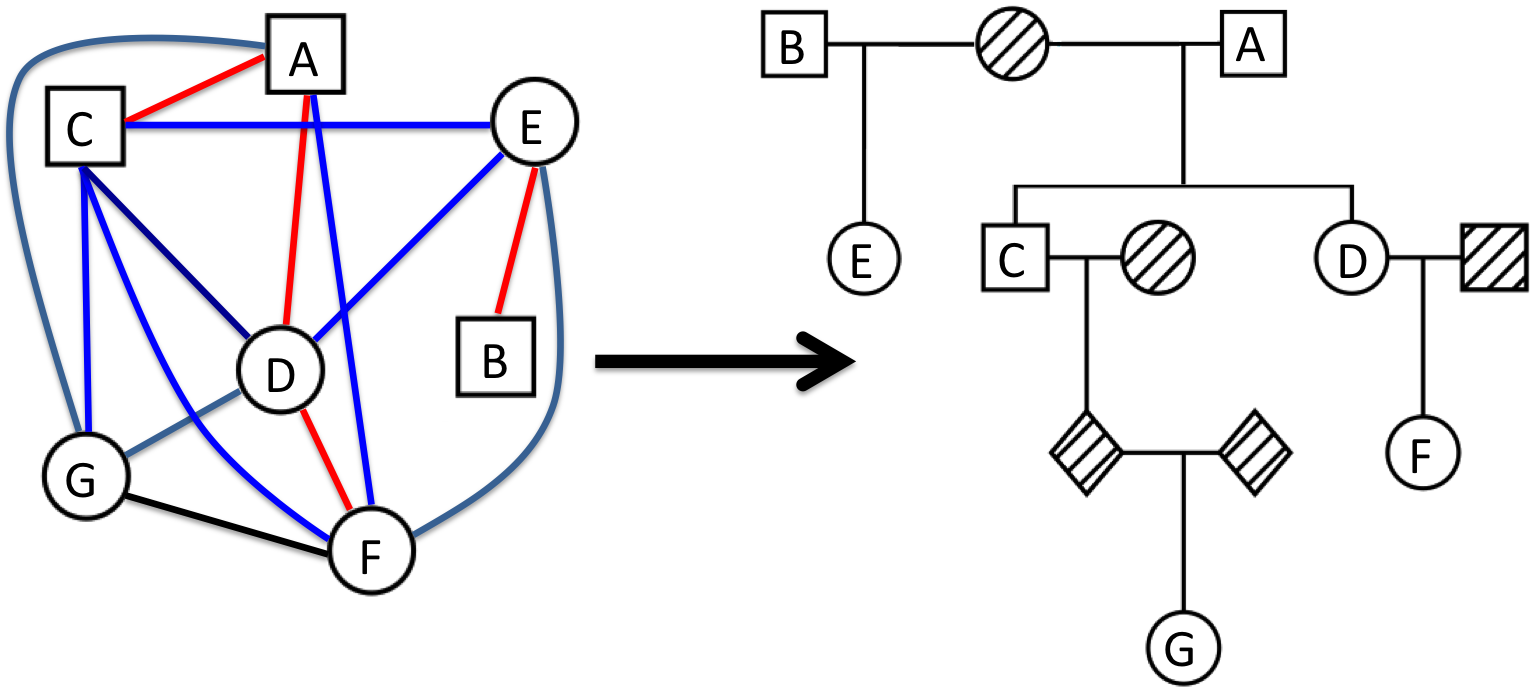
|
||||
Maximum Unrelated Set Identification (IMUS)
Jeffrey Staples, Deborah A. Nickerson, and Jennifer E. Below (2013). Utilizing graph theory to select the largest set of unrelated individuals for genetic analysis. Genet Epidemiol 37, 136-41.
The IMUS method is an algorithm adapted from graph theory that always identifies the maximum set of unrelated individuals in any dataset, and allows weighting parameters to be utilized in unrelated sample selection. PRIMUS reads in user-generated IBD estimates and outputs the maximum possible set of unrelated individuals, given a specified threshold of relatedness. Additional information for preferential selection of individuals may also be utilized. For example, when there are two equally sized maximum sets of unrelated individuals in a network, PRIMUS can preferentially select the set with more affected individuals. Primus can also preferentially select on quantitative traits such as data missingness of each sample.
Pedigree Reconstruction (PR)
Jeffrey Staples, Dandi Qiao, Michael H. Cho, Edwin K. Silverman, University of Washington Center for Mendelian Genomics, Deborah A. Nickerson, and Jennifer E. Below, PRIMUS: Rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. (in press). American Journal of Human Genetics.
The PR algorithm is a method to reconstruct pedigrees within a genetic dataset. PRIMUS can verify expected pedigree structures from genetic data, and it can identify and incorporate novel, cryptic relationships into pedigrees. Pedigree reconstruction can be ran by providing genetic data in the PLINK or PLINK2 input file formats and the prePRIMUS pipeline will attempt to generate IBD estimates that will be used for reconstruction. Alternatively, you can provide IBD estimates directly. PRIMUS uses the IBD estimates to first break the dataset into family networks and then reconstruct each network separately.
PRIMUS outputs the project level results (i.e., the results for the entire dataset) in the *_PRIMUS/ directory by default. Within the project level directory you should find two summary files, a directory for each family network, a *.dot file for networks containing 5 or more samples, and *unrelated_samples.txt file. The first project level summary file provides high-level statistics for the entire dataset and for each family network. The second project level summary file lists the pairwise relationships for all the samples in the project, which is generated using all possible pedigrees reconstructed for each family network. The *_unrelated_samples.txt is a list of all the samples that were not included in any family network. The *.dot file can be open with any program that can visualize .dot files, such as graphviz.
Each network directory contains a network summary file and files for each possible pedigree that fits the IBD estimates for that family network. The network summary file provides lower level statistics for the entire network as well as for each possible pedigree. The possible pedigrees will be ranked from most likely, 1, to least likely, N, where N is the number of possible pedigrees for that network. For each possible pedigree there is a *.fam file containing the relationships, a *.ps file that is an image of the pedigree generated by Cranefoot that can be opened by any program that can display post-script files, a *.genome file that contains the IBD estimates for only the samples in that family network, and a mz_twins file that list all pairs of MZ twins (or duplicate samples).
PRIMUS provides a lot of useful input options, including sex status, affection status, and samples' ages. PRIMUS will use the sex of each sample to improve reconstruction and will include the sex of the samples in the *.fam file. PrePRIMUS will automatically try to determine the sex of each sample if sex data is included in the input genotype file. Affection status will be included in the *.fam files as well as in the pedigree images (*.ps files). Age will be used to flag pedigrees that are inconsistent with the reported ages, e.g. if the child is older than the parent.
PRIMUS has two brand new input features (not yet published). First PRIMUS can use mitochondrial (MT) and non-recombining Y (NRY) data to improve reconstruction. If MT and NRY data are included in the PLINK formatted input file, then PRIMUS will attempt to use them to improve reconstruction. Alternatively, MT and NRY pairwise comparisons can be input as a text file if the correct command line options are used. Second, PRIMUS can input the results from ERSA in order to use the distant relationships predicted by ERSA to connect the family networks reconstructed by PRIMUS. This is particularly useful in large genetic dataset containing cryptic relatedness and in large, sparsely sampled families.
Licensing and Download
Software implementing the ERSA algorithm is now available. Register to download the most recent version here.
Previous Versions